Article Text

Abstract
Objective A predictable relation between genotype and disease expression is needed in order to use genetic testing for clinical decision-making in hypertrophic cardiomyopathy (HCM). The primary aims of this study were to examine the phenotypes associated with sarcomere protein (SP) gene mutations and test the hypothesis that variation in non-sarcomere genes modifies the phenotype.
Methods Unrelated and consecutive patients were clinically evaluated and prospectively followed in a specialist clinic. High-throughput sequencing was used to analyse 41 genes implicated in inherited cardiac conditions. Variants in SP and non-SP genes were tested for associations with phenotype and survival.
Results 874 patients (49.6±15.4 years, 67.8% men) were studied; likely disease-causing SP gene variants were detected in 383 (43.8%). Patients with SP variants were characterised by younger age and higher prevalence of family history of HCM, family history of sudden cardiac death, asymmetric septal hypertrophy, greater maximum LV wall thickness (all p values<0.0005) and an increased incidence of cardiovascular death (p=0.012). Similar associations were observed for individual SP genes. Patients with ANK2 variants had greater maximum wall thickness (p=0.0005). Associations at a lower level of significance were demonstrated with variation in other non-SP genes.
Conclusions Patients with HCM caused by rare SP variants differ with respect to age at presentation, family history of the disease, morphology and survival from patients without SP variants. Novel associations for SP genes are reported and, for the first time, we demonstrate possible influence of variation in non-SP genes associated with other forms of cardiomyopathy and arrhythmia syndromes on the clinical phenotype of HCM.
- GENETICS
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
Introduction
Hypertrophic cardiomyopathy (HCM) is a common autosomal dominant genetic trait associated with sudden cardiac death (SCD) and progressive heart failure.1 ,2 Patients are routinely offered genetic testing in order to provide them with information about the likely impact of disease on their lives and facilitate lifestyle and medical interventions that improve prognosis.2 ,3 However, for this strategy to succeed, there must be a predictable relation between specific genotypes and disease expression.
In around 50% of cases, HCM is caused by mutations in genes coding for sarcomere or sarcomere-related genes.4 So far, the most commonly reported genotype–phenotype associations are those that relate to the presence or absence of sarcomere protein (SP) gene mutations rather than mutations in specific genes.5 ,6 A number of studies have suggested that some mutations are associated with reduced survival, but these findings are inconsistent and fail to account for the often dramatic variation in clinical phenotypes seen in individuals with the same genetic variant.w1–w7 7 w8 8 w9 9 w10 w11 10 w12 11 w13 w14 12 ,13 w15–w18 14 w19 15 ,16
Several studies have examined the role of common genetic variation on the expression of sarcomere mutations using genome-wide association studies or a candidate gene approach, but most have failed to show any major effect on disease expression.w20–w22 17 ,18 HCM cases (as well as controls) also carry rare variants in genes coding for desmosomal, ion-channel and other proteins implicated in inherited heart disease19 but their relevance to disease expression is unknown.
The hypothesis of this study is that rare variants in sarcomere genes and also in non-sarcomere genes implicated in other forms of inherited cardiac disorders (for which sequence data are available in our study) modify the clinical characteristics and severity of HCM.
Methods
Study population and design, clinical evaluation and sample collection
The study was approved by the University College London (UCL)/UCL Hospitals (UCLH) Joint Research Ethics Committee. Before enrolment, all patients provided written informed consent and received genetic counselling in accordance with international guidelines.3
An observational, retrospective, longitudinal cohort study design was used. The study population comprised unrelated and consecutively evaluated patients with HCM referred to the Inherited Cardiovascular Disease Unit at The Heart Hospital, UCLH, London, UK. Clinical evaluation included a personal and family history, physical examination, 12 lead ECG, echocardiography, symptom limited upright exercise testing with simultaneous respiratory gas analysis (cardiopulmonary exercise test) and ambulatory ECG monitoring as previously described.20 HCM was diagnosed in probands when the maximum left ventricular (LV) wall thickness (MLVWT) on 2D echocardiography measured 15 mm or more in at least one myocardial segment or when MLVWT exceeded 2 SDs corrected for age, size and gender in the absence of other diseases that could explain the hypertrophy.21 In individuals with unequivocal disease in a first degree relative, diagnosis was made using extended familial criteria for HCM.22 Ethnicity was self-reported and classified using a modified National Health Service ethnic categorisation. Patients were evaluated every 6–12 months or earlier if there was a clinical event. Initial evaluation and follow-up data were collected prospectively and registered in a relational database. The definitions of severe LV hypertrophy, family history of SCD, syncope, non-sustained ventricular tachycardia (NSVT) and abnormal blood pressure response were as previously described.23
Targeted gene enrichment and high-throughput sequencing
Blood samples were collected at initial evaluation and DNA was isolated from peripheral blood lymphocytes using standard methods. The sequencing methodology has been reported in detail previously.19 In summary, the protocol was designed to screen 2.1 Mbp of genomic DNA sequence per patient, covering coding, intronic and selected regulatory regions of 20 genes known to be associated with HCM and dilated cardiomyopathy (MYH7, MYBPC3, TNNT2, TNNI3, MYL2, MYL3, ACTC1, TPM1, TNNC1, MYH6, CSRP3, DES, TCAP, PDLIM3, PLN, LDB3, LMNA, VCL, RBM20 and TTN), 10 genes implicated in arrhythmia syndromes/ion-channel disease (RYR2, KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2, ANK2, CASQ2, CAV3 and KCNJ2), seven genes associated with arrhythmogenic right ventricular cardiomyopathy (PKP2, DSC2, DSG2, JUP, DSP, TMEM43 and TGFß3) and a further four candidate genes (GJA1, PLEC, PNN and PKP4) which were not analysed in this work.19 Analysis of titin (TTN) variants and their effect on phenotype is ongoing and will be reported in a separate paper.
Bioinformatic analysis
Paired-end reads were aligned using Novoalign software V.2.7.19 on the human reference genome build hg19. Duplicate reads were flagged using the Picard MarkDuplicate tool. Our calling strategy followed closely the Genome Analysis Toolkit (GATK) best practices as of January 2014. Briefly, following BAM file compression using the GATK ReduceReads module,24 multisample calling was performed on all probands jointly with a set of 1492 unrelated whole exomes (UCL-exome consortium) using the GATK Unified Genotyper.24 After GATK variant recalibration (separately for SNPs and indels), calls were annotated using the ANNOVAR software (with the Ensembl gene definitions).w23 For all association tests, we filtered variants for the GATK recalibration PASS filter.
Candidate variants for further analysis were defined using frequency and predicted functional effect. For the functional filter, exonic non-synonymous, loss-of-function and splice-site variants were included. Sequence data were filtered using a minor allele frequency threshold of ≤0.2% based on the NHLBI exome variant server data (computed through the ANNOVAR annotations). To provide a more accurate estimate of variant frequency in controls that is not affected by potential differences in calling strategy in the NHLBI dataset, we randomly selected 25% of the 1492 UCL-exome samples as an ‘external control set’ and removed variants that appeared more than twice in these 372 ‘external controls’. These samples were only used to define a variant frequency and not included in the subsequent association test, to avoid a previously noted statistical issue, where variant frequency is defined in the same set that is used for case control testing.w24 Variants present in the dbSNP build 137 databasew25 and published in the literature were identified. In silico prediction of pathogenicity for novel missense variants was performed using Polyphen2, SIFT and Condel.w26 w27 25 A variant was predicted to be pathogenic if classified as ‘damaging’ by SIFT and simultaneously ‘possibly’ or ‘probably damaging’ by Polyphen2, or if predicted to be damaging by Condel.
Summary statistics for genotype–phenotype associations
R (V.3.0.0) and SPSS (V.22.0.0.0, IBM Corp.) were used for the analyses. Clinical phenotype data are presented as frequency (and percentage) for non-continuous variables and mean±SD or median and IQR for continuous variables where appropriate. Normally distributed continuous variables were compared using unpaired two-tailed Student's t test. Multiple groups were compared using analysis of variance. Categorical variables were compared using χ2 or Fisher exact tests. When appropriate, non-parametric tests were used.
Group comparisons were made for the prevalence and severity of each phenotypic trait (at baseline and final follow-up) in patients with and without a rare variant in one or more of the eight most common SP genes (MYH7, MYBPC3, TNNI3, TNNT2, MYL2, MYL3, ACTC1 and TPM1). We also compared the prevalence and severity of each phenotypic trait in patients carrying only one versus more than one variant in SP genes. The same comparisons were made for the presence and absence of rare variation in non-SP genes in the whole cohort and in the subgroup of individuals with a disease-causing SP gene mutation.
Multiple testing correction strategy
For each trait of interest we tested the effect of variants in eight SP genes and 28 non-SP genes. Therefore, a nominal p value of 0.05 was not appropriate. In addition, the Bonferroni correction for the number of phenotypes multiplied by the number of genes is too stringent because it tests the global null of no association between any pair of gene/trait. We therefore took an intermediate approach, correcting the analysis of each phenotype for the number of gene tests. For SP genes, we performed nine tests (one per SP gene, plus an additional test for all SP mutations combined). Therefore, we used p<0.0056, which is 0.05/9. For non-SP genes, we corrected for 28 tests, which translates into p<0.0018=0.05/28. Data on associations that did not fulfil these thresholds but met a nominal p value of <0.05 are presented in the Results section and online supplementary files.
Survival analysis
Definition of endpoints in the survival analyses was as previously described.23 Survival from cardiovascular death (a composite of SCD and death from heart failure or stroke) and SCD or equivalent (appropriate implantable cardioverter-defibrillator (ICD) shock) was modelled using Kaplan–Meier analysis and log-rank test from the first clinical evaluation at The Heart Hospital and from birth.
Results
Study population
In all, 874 unrelated and consecutive patients with HCM were studied. Mean follow-up time was 4.8±3.5 years (0–16.8 years). Table 1 summarises the demographic and clinical characteristics of the patients at initial evaluation and their outcomes.
Demographic and clinical characteristics of the study cohort
SP genes variants
Overall, 383 patients (43.8%) had 265 distinct rare (minor allele frequency ≤0.2%) variants in one or more the eight SP genes most commonly associated with HCM (MYH7, MYBPC3, TNNT2, TNNI3, MYL2, MYL3, ACTC1 and TPM1) (table 2 and see online supplementary table S1). A total of 142 (53.5%) of these rare variants were published previously as disease-causing mutations; 44 (16.6%) were novel missense variants predicted in silico to be pathogenic and 40 (15%) were novel potential loss-of-function variants. In all, 37 patients (4.2%) carried multiple candidate variants in these eight SP genes.
Prevalence of rare variants (minor allele frequency ≤0.2%) in the eight main sarcomere genes
Non-SP gene variants
In all, 114 distinct rare desmosomal protein gene variants were present in 122 (14.0%) patients; 192 rare ion-channel disease gene variants were present in 196 patients (22.4%). A total of 29 (25.4%) of the desmosomal variants and 38 (19.8%) of the ion-channel variants were published previously. A further 74 (24.2%) of these non-sarcomere variants were novel missense variants predicted in silico to be pathogenic and 20 (6.5%) were potential loss-of-function variants. In all, 122 patients (43.0% of 284) with these non-SP variants also carried a SP variant.
Genotype–phenotype associations
Genotype–phenotype associations significant at the defined stringent thresholds are summarised in figures 1⇓–3 and table 3. A complete list of p values significant at p<0.05 for all pairs of traits/genes is provided in table 4 for non-SP genes and online supplementary table S2 for SP and related genes. Online supplementary table S3 summarises the associations for non-SP genes presented in tables 3 and 4, analysed within the subcohort of sarcomere-positive individuals only.

Comparison between sarcomere gene mutation-positive and -negative patients. (A) Age at initial evaluation (45.78±14.65 vs 53.05±14.94 years). (B) Family history of hypertrophic cardiomyopathy (HCM). (C) Family history of sudden cardiac death (SCD). (D) Hypertrophy pattern. (E) Maximum wall thickness (18.83±4.42 vs 18.12±4.08 mm). (F) Implanted implantable cardioverter-defibrillators (ICDs). Key: 0: sarcomere-negative; 1: sarcomere-positive. For B, C, E: red colour and percentages indicate the individuals with the trait within each genotype; for D light blue—asymmetric septal hypertrophy; red—apical hypertrophy; green—concentric hypertrophy.
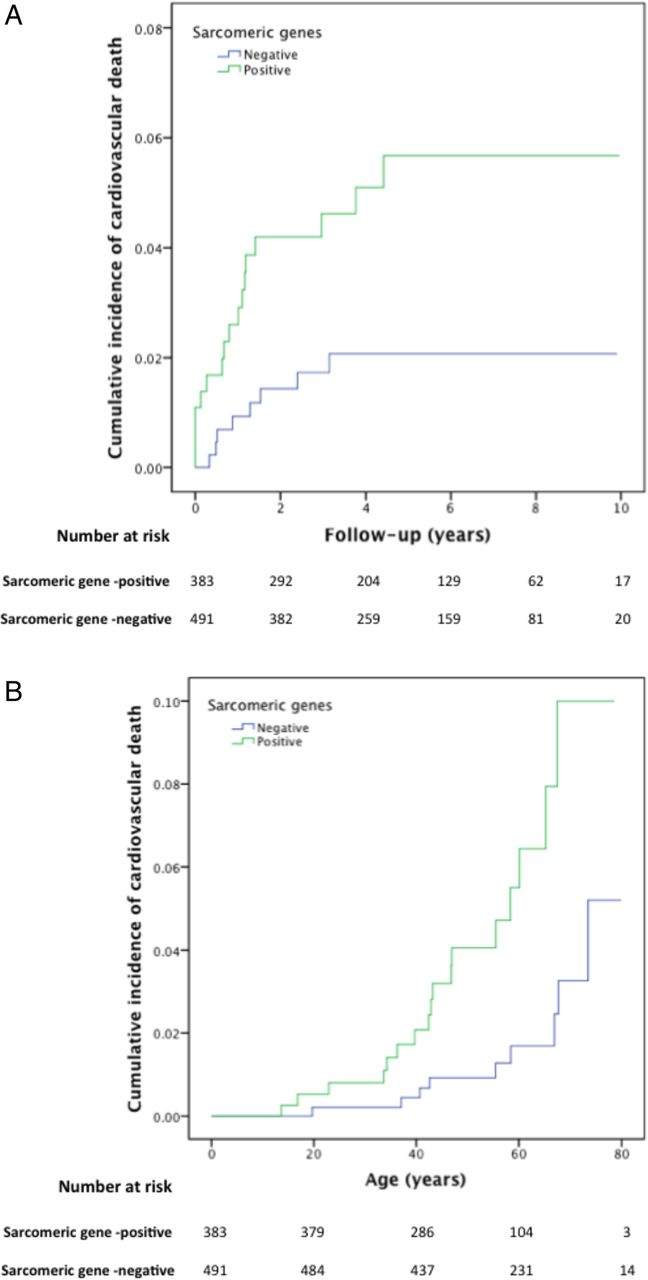
Kaplan–Meier cumulative incidence curves for cardiovascular death (see Methods section), comparing sarcomere-positive and sarcomere-negative individuals, modelled for (A): follow-up from first evaluation (years), log-rank test p value=0.012 (HR 2.81; 95% CI 1.21 to 6.51) and (B): time from birth (years), log-rank test p value=0.001 (HR 3.99; 95% CI 1.71 to 9.36). The Y axis values indicate proportions.
{kind=link}
{kind=link}
{kind=link}
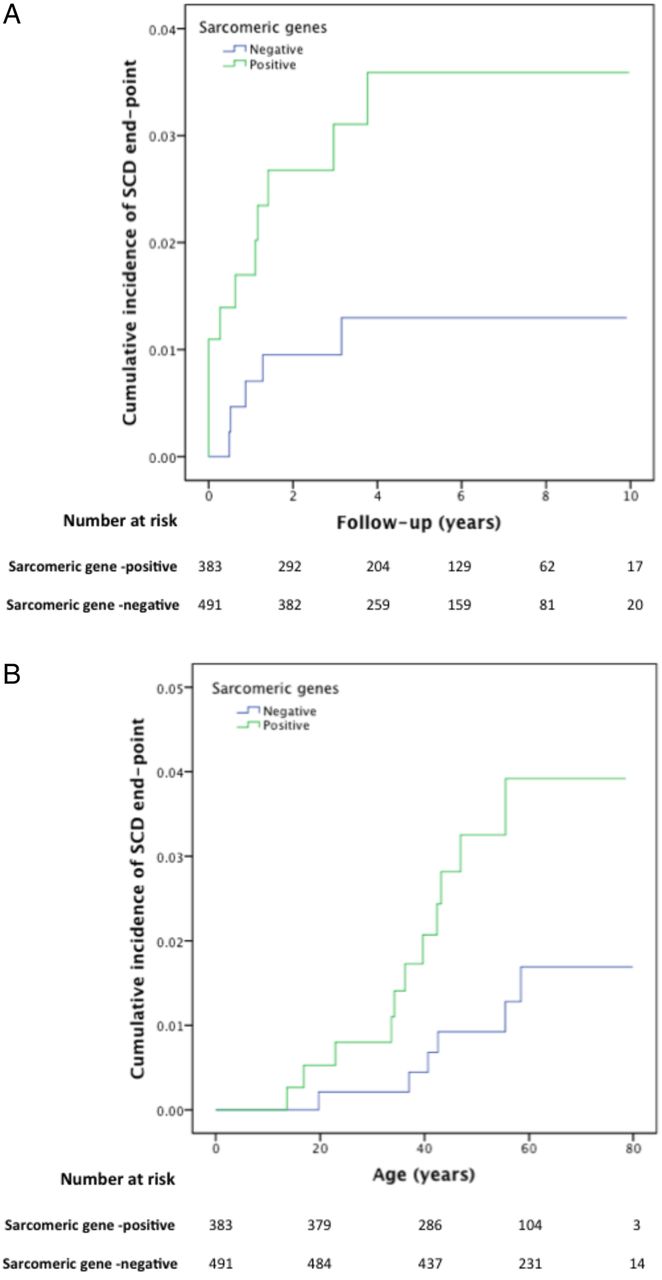
Kaplan–Meier cumulative incidence curves for sudden cardiac death/aborted sudden cardiac death, comparing sarcomere-positive and sarcomere-negative individuals, modelled for (A): follow-up from first evaluation (years), log-rank test p value=0.039 (HR 2.89; 95% CI 1.01 to 8.33) and (B): time from birth (years), log-rank test p value=0.028 (HR 3.44; 95% CI 1.19 to 9.92) . The Y axis values indicate proportions.
Genotype–phenotype associations for individual sarcomeric and related protein genes and non-sarcomere protein (SP) genes meeting the predefined statistical thresholds for multiple testing.
Genotype–phenotype associations for non-sarcomeric protein genes not meeting the predefined statistical thresholds for multiple testing
Effect of mutations in sarcomere genes
Patients with at least one variant in one of the eight main sarcomere genes were younger at diagnosis and had a higher frequency of a family history of HCM or SCD compared with those without sarcomere variants. Patients with SP mutations were more likely to have asymmetric septal hypertrophy than apical or concentric patterns and had greater MLVWT. The prevalence of male sex was lower in sarcomere-positive individuals (62.4% vs 72.0%, p=0.00213); these individuals were also more likely to have an ICD implanted. Patients with sarcomere mutations had a lower resting systolic blood pressure (SBP) (123.1±19.2 vs 133.7±21.3 mm Hg, p=1.54×10−9) and a lower SBP response to exercise (44.1±21.5 vs 52.2±26.9 mm Hg, p=7.61×10−5).
Similar and additional associations were observed when individual SP genes were considered (table 3 and see online supplementary table S2).
The proportion of cardiovascular deaths during follow-up was higher in patients with at least one variant in one of the eight main SP genes. The same was true for sudden death/ICD discharge (figure 2 and 3).
Patients with multiple SP gene variants
Patients who carried more than one sarcomere variant had an increased prevalence of syncope when compared with individuals with only one sarcomere variant (35.1% vs 16.6%; 13/37 vs 56/337, p=0.012). SBP response to exercise was lower in individuals with multiple sarcomere variants compared with a single variant (36.5±21.9 vs 45.1±21.2 mm Hg, p=0.012) and there was a higher proportion of patients with an abnormal blood pressure response to exercise (10/29 vs 39/276; 34.5% vs 14.1%, p=0.010).
Associations with rare variants in desmosomal and ion-channel genes
A total of 71 patients carried rare ANK2 variants (of these, 36 also carried SP variants). At a significance threshold of p<0.0018, the proportion of patients with an MLVWT ≥30 mm was greater in carriers of an ANK2 rare variant (table 3). This association was still present when restraining the analyses to the subcohort of sarcomere-positive individuals only (see online supplementary table S3).
Additional genotype–phenotype correlations were identified at a less stringent p<0.05. These are listed in table 4, and include an increased mean MLVWT in ANK2 variant carriers.
Discussion
In this study of a large consecutive cohort of HCM probands screened with high-throughput sequencing, we have detected a class effect of SP gene variants on the HCM phenotype and identified novel associations with mutations in individual SP genes. We also demonstrated evidence of an association between non-SP genes and disease expression that could explain some of the characteristic clinical heterogeneity of HCM.
Influence of sarcomeric variation on phenotype
The presence of any sarcomere variant was associated with an asymmetric septal hypertrophy pattern, younger age at presentation, family history of HCM and SCD and female gender. This study also shows that patients with SP variants had higher cardiovascular and sudden death-related mortality during follow-up. Patients with more than one SP variant had more SCD risk markers, consistent with the suggestion in previously published series of a gene dose effect.13 ,26–28 However, the low number of outcome events during follow-up may have biased the survival analysis and precluded an analysis of other associations, including the effect of carrying multiple compared with single variants. The survival from birth is provided for comparison with the published literature but also introduces an inherent survivor bias. With regard to individual SP genes, we demonstrate a number of novel associations that provide evidence for mutation specific effects on clinical phenotype and prognosis.
Modifier effect of non-sarcomere variants
The data in this study suggest that rare ANK2 variants are associated with severe LV hypertrophy. ANK2, or ankyrin B, stabilises membrane ion-channels in cardiomyocytes and mutations in the gene cause long QT syndrome 4, ventricular arrhythmias and sinus node disease.29 ,30 We are unaware of any link between ANK2 expression and changes in LV morphology, but as ankyrins interact with proteins that influence calcium homeostasis and ß-adrenergic signalling, it is conceivable that they eventually affect the cellular phenotype that results from a primary SP gene variant. The strength of the statistical association (p=0.0005) exceeds the requirement of a Bonferroni correction for the number of tested genes (36 independent tests), but further replication in independent cohorts will be necessary to confirm these results.
In addition to the association with ANK2 variation, we detected a number of associations at lower statistical significance with variation in other non-SP genes. Patients with SCN5A rare variants were more likely to have left atria enlargement at their last evaluation. A link between SCN5A disruption and TGF-β1-mediated fibrosis has recently been demonstrated in a murine model of sinus node diseasew28 and it is possible that SCN5A variants influence the pro-fibrotic milieu associated with SP mutations. SCN5A rare variation was also associated with a higher proportion of LV outflow tract obstruction. Individuals with PLN rare variants were more likely to have NSVT, which is interesting considering the recently described arrhythmogenic risk of a founder PLN mutation.w29 As for the association with ANK2, replication of these findings is required.
Clinical implications
If genetic variation is to become a clinically relevant biomarker, it is essential that there is a clear understanding of genotype–phenotype relationships. The associations between sarcomere gene variants and the broad phenotype examined in this study contribute to this understanding and, if confirmed in other populations, could inform the counselling of patients and relatives who are contemplating predictive genetic testing. The demonstration that non-sarcomeric variants may influence disease expression is an illustration of the complexity that underlies the biology of this disease. New models that incorporate a broad genetic profile and deep clinical phenotyping are necessary to test the role of mutation analysis in prognostic models.
Key messages
What is known on this subject?
In up to 50% of cases, hypertrophic cardiomyopathy is caused by mutations in genes coding for sarcomere or sarcomere-related genes, but the often dramatic variation in clinical phenotypes caused by the same or similar mutations remains largely unexplained.
What might this study add?
This study presents novel genotype–phenotype associations in a large cohort of 874 patients using high-throughput genetic sequencing. We describe a strong class effect for sarcomeric protein variants on clinical presentation, LV morphology and survival. For the first time, a modifier effect of rare variants in non-sarcomeric genes associated with other forms of cardiomyopathy and arrhythmia syndromes is demonstrated.
How might this impact on clinical practice?
These are novel findings which suggest new and testable insights on the biology and pathophysiology of the disease that might eventually have important clinical implications for counselling of patients and risk prediction models.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online references
- Data supplement 2 - Online table 1
- Data supplement 3 - Online table 2
- Data supplement 4 - Online table 3
Footnotes
Correction notice The license of this article has changed since publication to CC BY 4.0.
Contributors LRL: conception and design of the study, analysis and interpretation of data, drafting of the manuscript, and final approval of the manuscript submitted; PS, WJM, PME and VP: conception and design of the study, analysis and interpretation of data, revising the manuscript critically for important intellectual content, and final approval of the manuscript submitted; OPG, MH, CO’M, HCT, CD, SJ and LM: analysis and interpretation of data, revising the manuscript critically for important intellectual content, and final approval of the manuscript submitted.
Funding LRL was supported by a grant from the Gulbenkian Doctoral Programme for Advanced Medical Education, sponsored by Fundação Calouste Gulbenkian, Fundação Champalimaud, Ministério da Saúde and Fundação para a Ciência e Tecnologia, Portugal. OPG received research support from the British Heart Foundation. This work, including support for Chrysoula Dalageorgou, was undertaken at UCLH/UCL who received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. LM received funding from the grant: FIS 2011: PI11/02604. Instituto de Salud Carlos III, Madrid, Spain. VP is partly supported by the National Institute of Health Research (NIHR) Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology.
Competing interests LM is a shareholder of Health in Code SL.
Ethics approval University College London (UCL)/UCL Hospitals (UCLH) Joint Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Heartbeat