Article Text

Abstract
OBJECTIVE To determine the role of healed plaque disruption in the generation of chronic high grade coronary stenosis.
METHODS Coronary arteries obtained at necropsy were perfuse fixed with formal saline for 24 hours at 100 mg Hg. The percentage lumen diameter stenosis was measured in each 3 mm segment containing a plaque, using the lumen size at the nearest histologically normal segment as the reference point. Each segment was prepared for histological examination and stained with Sirius red and immunohistochemistry for smooth muscle actin. Healed disruption was considered to be present when under polarised light there was a break in the yellow–white dense collagen of the cap filled in by more loosely arranged green collagen. Increased smooth muscle density in the green staining areas was required. Each section was read independently by two observers; any segment with discordant views was considered negative.
MATERIAL 31 men aged 51–69 dying suddenly of ischaemic heart disease. 39 coronary arteries were studied containing 256 separate plaques, after excluding coronary arteries with old total occlusions, an acute culprit thrombotic lesion, diffuse disease without normal arterial segments, and arteries related to old myocardial scars.
RESULTS 16 of 99 plaques causing < 20% diameter stenosis had prior disruption. In the 21–50% stenosis range 16 of 86 plaques showed healed disruption. Stenosis ⩾ 51% by diameter was present in 71 plaques, 52 of which showed a healed disruption pattern. The difference between stenosis < 50% and stenosis ⩾ 51% was significant by the χ2test (p < 0.001).
CONCLUSIONS Subclinical episodes of plaque disruption followed by healing are a stimulus to plaque growth that occurs suddenly and is a major factor in causing chronic high grade coronary stenosis. This mechanism would explain the phasic rather than linear progression of coronary disease observed in angiograms carried out annually in patients with chronic ischaemic heart disease.
- atherosclerosis
- stenosis
- plaque disruption
Statistics from Altmetric.com
Until coronary arteriography came into widespread use the development of stable angina and even acute myocardial infarction was thought to be caused by a slow progressive linear growth of an atherosclerotic plaque. Angiography at regular intervals in patients with chronic stable ischaemic heart disease however1-3 showed that progression was phasic rather than linear with time. The occlusive lesions responsible for acute myocardial infarction often developed in arterial segments that were normal or mildly irregular in the previous angiogram.4 ,5 New angiographic lesions, often with high grades of stenosis, developed between annual angiography. While some high grade stenoses did progress slowly to occlusion many mild lesions remained static. Chronic total occlusion in the absence of any evidence of an episode of myocardial infarction appeared between angiograms in arterial segments which were not more than irregular in outline. The message of these studies was that intermittent, rapid episodes of plaque growth were responsible for much of the angiographic progression of coronary atherosclerosis.
Acute ischaemic episodes such as myocardial infarction and unstable angina have been shown by histology,6angiography,7 and angioscopy8 to be largely caused by thrombosis developing over a culprit plaque. The most common trigger to such episodes of thrombosis is plaque disruption. The initial view, largely based on necropsy studies, was that episodes of plaque disruption and thrombosis were always major events associated with clinical symptoms. This view changed when angioscopy and atherectomy studies compared the coronary lesions responsible for stable and unstable angina.9-11 The consistent message emerged that a small proportion of plaques responsible for stable angina had residual thrombus. Pathology studies that looked in more detail at the coronary arteries of subjects dying with unstable angina and myocardial infarction12 found that there was one main culprit disrupted plaque, but often individual patients had other minor subsidiary thrombotic events in other sites in the coronary artery tree.13 ,14 In 130 subjects dying suddenly of ischaemic heart disease, 314 episodes of coronary thrombosis were found: an average of 2.4/patient. In subjects who had coronary atheroma but who died suddenly of obvious non-cardiac causes, 14% had histological evidence of small recent disruption and thrombosis episodes based on finding recent thrombus in or on the plaque.15 ,16 These findings suggest that episodes of disruption and minor thrombosis are an integral part of atherosclerotic disease and have the potential to cause sudden plaque growth in the absence of any clinically recognised acute ischaemic event.
Many patients survive episodes of both clinically expressed and silent plaque disruption. The healing process at the site has been studied in human subjects16 and experimental models using mechanical injury to the vessel wall.17 Thrombus is initially lysed to a greater or lesser extent, then smooth muscle cell proliferation develops to synthesise new collagen that restores the integrity of the injured intima. Finally, re-endothelisation occurs, although long before this any exposed connective tissue is passivied by the binding of proteoglycans to become inert to platelets. The newly proliferating smooth muscle cells have a characteristic criss–cross storiform arrangement often designated as accelerated healing.18 The final result of the healing of an episode of disruption may range from little increase to high grade stenosis.
It is uncertain whether an episode of healed disruption and thrombosis is responsible for a small or large proportion of high grade stenoses in human coronary disease. It is unclear whether pure atherosclerotic plaque growth without any thrombotic component ever causes stenosis. These questions can only be answered if there were a morphological method of recognising healed disruption. The recently laid down collagen in the repair process has a different staining pattern and fibre organisation than the original collagen of the plaque16 when viewed under polarised light after staining with Sirius red.19 This technique can be used to determine what proportion of high grade coronary stenotic lesions have had an episode of disruption.
Materials and methods
Thirty one consecutive men aged 51–69 dying suddenly of ischaemic heart disease within six hours of the onset of any symptoms were studied in detail at necropsy. None of the 31 had any cause of death other than coronary artery disease with a culprit thrombotic lesion. Fifteen had a prior history of infarction or stable angina, 13 of whom had received treatment for hypertension. Five of the 31 were diabetic.
In all cases the coronary arteries were perfuse fixed with 10% formal saline at a pressure of 100 mg Hg from the aorta against the closed aortic valve for 24 hours. The coronary arteries were dissected intact from the heart and decalcified. Thirty nine portions of artery 5 cm long were studied. After decalcification the artery was cut into transverse sections at 3 mm consecutive intervals. The cross sectional area of the distended lumen was measured at each segment by a video camera attached to an AMS Optomax V image analysis system. Each transverse section was then prepared for histological examination and stained by Sirius red and immunohistochemistry for smooth muscle actin. All the arteries examined had discrete areas of stenosis interspersed with segments of artery confirmed by histology to be normal. Arteries with culprit lesions, chronic total occlusions, and confluent stenoses narrowing the whole arterial segments were examined. Arterial segments related to regional myocardial scars were excluded. Each subject contributed at least one artery for study. The cross sectional area of the artery lumen was used to construct a linear plot from which the percentage diameter stenosis was calculated at each plaque site using the adjacent histologically normal segment as the reference point.
CRITERIA FOR PRIOR DISRUPTION
The criteria used were a discrete defect in the plaque cap collagen (yellow) filled by collagen staining green in the Sirius red stain viewed under polarised light (fig 1) followed by analysis of the image to pseudocolour (fig 2A). The area had to contain an increased density of smooth muscle cells to be regarded as positive (fig 2B). Two observers read the slides independently. Where there was disagreement the segment was regarded as not showing disruption.

A cross section of a healed disruption in a human coronary plaque stained by Sirius red and viewed under polarised light. Much of the plaque collagen is yellow–white. A residual lipid core is present. Within the plaque cap there is a large defect filled with more loosely arranged green staining collagen.

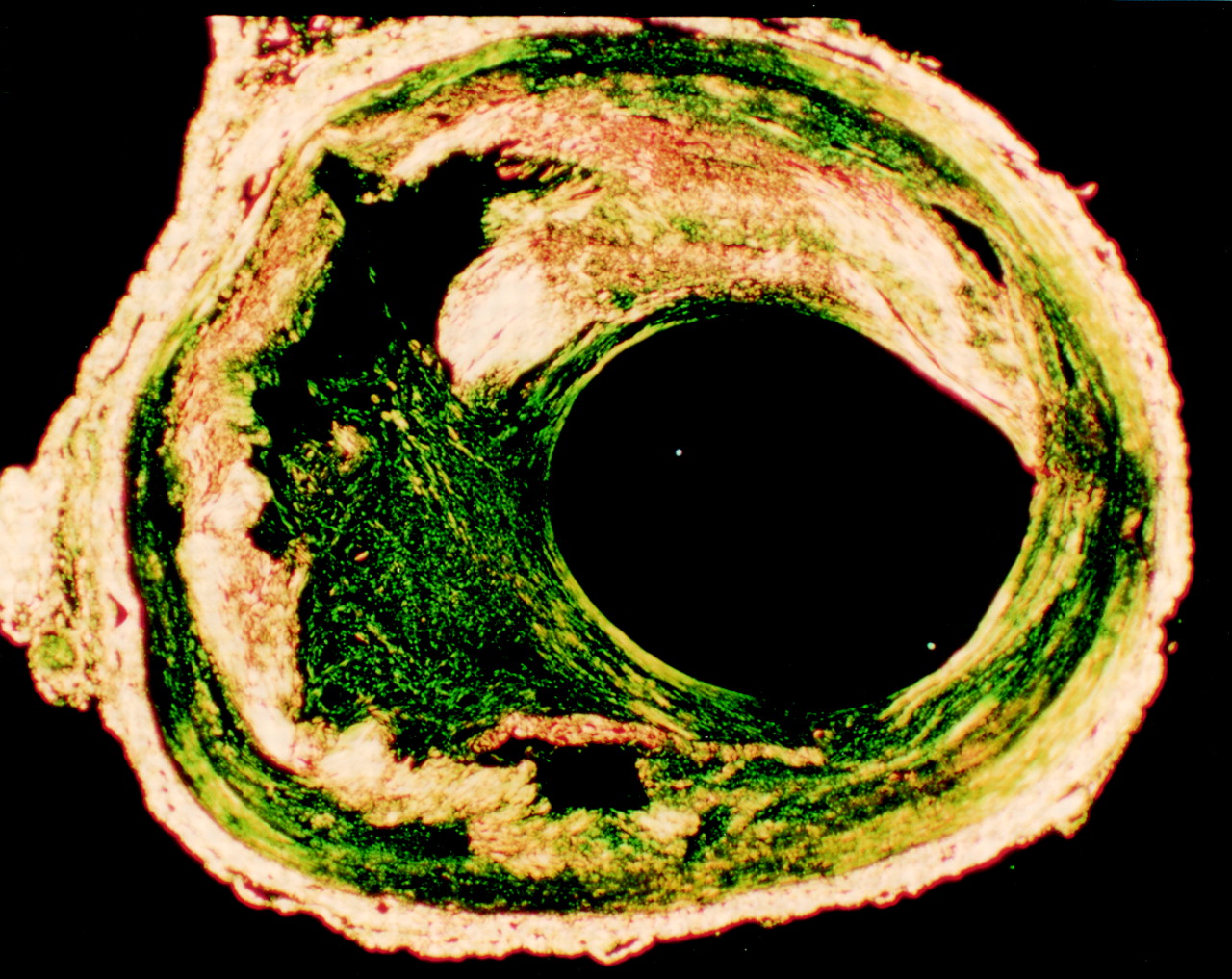{kind=link}
{kind=link}
(A) Using image analysis and colour segmentation a computer image can be produced in which the dense older collagen is magenta in colour and the more loosely arranged collagen green (L, lumen; C, core). The area of healing in the cap (arrow) is clearly delineated. (B) Immunohistochemistry for smooth muscle actin (red). The media stains completely due to its smooth muscle content. In the area of healing of the plaque cap (arrow) there is an increased concentrate of smooth muscle cells.
Results
Two hundred and fifty six separate plaques were studied. With diameter stenosis of 0–20% 16 of 99 plaques (16.2%) showed prior healed disruption. Stenosis of 21–50% diameter was present in 86 separate plaques of which 16 (18.6%) showed a healed disruption pattern. Fifty two of 71 plaques (73.2%) with stenosis ⩾ 51% in diameter had healed disruption patterns. The difference between stenosis < 50% and ⩾ 51% was highly significant using the χ2 test (p < 0.001). In 12 arterial segments related to regional infarcts used as controls, nine had images indicating the technique would identify prior disruption episodes.
Discussion
There is seldom a close agreement between pathology measurements of coronary stenosis and angiographic measurements.20 ,21The method used here (perfusing the arterial lumen at a pressure sufficient to achieve a round lumen equivalent of maximal vasodilatation before any shrinkage introduced by tissue processing for histology) means that the methods of measuring stenosis are exactly equivalent to those used in angiography in vivo.22 The diameter of the test segment was compared to the diameter of the nearest arterial segment where the wall was histologically normal. In this study, therefore, > 50% diameter stenosis is what the clinician would regard as significant in life.
The results show that at least 70% of high grade stenotic lesions have had an episode of prior disruption as the trigger to enhanced accelerated smooth muscle proliferation causing an increase in plaque volume. The figure is likely to be an under rather than an overestimate. Plaque disruption more remote in time could become unrecognisable when the collagen becomes more dense. There is no way of knowing whether this takes one or two years or even longer. Even allowing for an error rate that underestimates the frequency of plaque disruption, there are some plaques causing high grade stenosis that do not have a structure suggesting they had any disruption in the past. They retain a uniform plaque cap and a lipid core. It cannot be excluded that thrombus deposited on the surface of these plaques due to endothelial erosion played a part in the growth in the past. There does however appear to be a small population of plaques where simple plaque growth by lipid accumulation and production of collagen by smooth muscle cells has occurred—the growth of this population of plaques could be linear. The study was designated to consider the role of healed disruption in the genesis of discrete stenotic segments in which there were adjacent normal segments of artery. Diffuse stenosis involving long segments of artery were not studied and may have a different pathogenesis. The highly significant difference between stenosis of < 50% compared with ⩾ 51% could be interpreted in two ways. One explanation is that high grade stenosis is complicated by disruption rather than caused by it. The evolution of lesions that cause acute infarction, very largely due to disruption, however shows that they are not previously stenotic23 and makes the explanation that disruption leads to stenosis far more tenable.
The concept that episodic plaque growth is triggered by subclinical thrombotic/disruption/erosion episodes would be in accord with the observed pattern of angiographic progression in coronary artery disease. It has the implication that an intervention such as lipid lowering, which reduces acute event rates, probably in large part due to stabilising the plaque and reducing the risk of clinically expressed disruption, would also reduce the risk of subclinical events and thereby slow angiographic progression. Angiographic progression and the risk of an acute event are known to be linked.24 ,25
The generation of high grade stenosis is dependent on another process in addition to an increase in plaque bulk. As a plaque evolves the artery undergoes local remodelling to increase its cross sectional area thus preserving normal lumen dimensions.26 This compensatory dilatation may be adequate to prevent stenosis formation but in other situations fails and stenosis develops.27 ,28Success or failure of remodelling appears to be plaque rather than patient specific and is a major factor in determining whether stenosis develops. One explanation for a failure of remodelling may be that plaque growth stimulated by disruption is too rapid for compensatory changes to develop in the arterial wall.